Help using the database – v0.7a
FAQ
Families
Motifs
Alignments
Phylogeny
Structure
Frequently asked questions
- Why another kinase database? There are several excellent resources on protein kinases, including kinase.com and the Kannan lab protein kinase ontology server. Both of these resources assume a fair degree of expertise with kinase terminology, and neither are focused on infectious disease. In addition, neither allow easy access to the underlying alignments, nor allow searching based on specific positions. Our goal with this database is to provide a user-friendly resource useful for both kinase experts and the parasitology community. Suggestions to this end are welcome, especially while we are in our alpha- and beta- stages.
- How can this database help me? If you aren't already an expert on kinases, check out the overview of kinase structure and nomenclature. Both family pages and individual kinase pages are annotated with a precis of published information, when available. Downloadable sequence logos are generated using the Bloom lab's dmslogo python package. We have created a custom alignment viewer and an in-browser phylogenetic tree viewer to facilitate interaction with the evolutionary relationships between the kinases in the database. Kinase structures can also be viewed in-browser using the LiteMol plugin.
- I'm interested kinase inhibitors – what have you got for me? The motifs search feature allows you to screen the kinomes of all organisms in the database for specific residues at a combination of given positions (e.g. the gatekeeper), including custom motifs that you define. For instance, if you've got a new compound developed for one kinase, this can help you identify kinases from pathogens that may also be targets. In the future, if there is community interest, we may attempt to create a repository of binding/inhibitory information for different molecules. Let us know if you've suggestions on how to accomplish such a goal!
- This is great, but I'd really like to see data from organism X... We are interested in expanding the database, and are happy to take suggestions. Please email Dr. Michael Reese with any constructive criticism about the site (or notable bugs!).
- I've found a bug (or problem with annotation)! What should I do? Please report bugs to Michael Reese. Screenshots, any information output to the browser console, and information how to replicate it would be most appreciated.
- Have you got anything more planned? The site is currently in an alpha-state. We expect our full v1.0 release to have high-quality alignments for the protein kinases from all major Apicomplexan clades and representative host organisms, as well as from outgroups including green and red algae and ciliates. In the near future, we hope to expand the database to include other eukaryotic pathogens – we take requests!
We hope to also include high-quality homology models for all kinases for which there is no structural information, but for which
reliable homology models can be created. Time permitting, we hope to allow some additional manipulation in-browser
(e.g. superposition of two structures).
Exploring kinase families

The database family explorer allows the facile browsing of the kinase families available in the database.
Kinases have been organized into "Superfamilies," "Families," and in some cases, "Subfamilies." The browser gives an overview of the members,
and autogenerated sequence logos of the major canonical motifs. A filtered
list of the members can be viewed in the resulting table, and subsets of these kinases can be selected using the checkboxes in the leftmost table
column. Such selections can be loaded into the motif browser, used to generate sequence logos
of the subset, or loaded into the multiple sequence alignment viewer or the
phylogenetic tree viewing tool.
Exploring kinases by conserved motifs
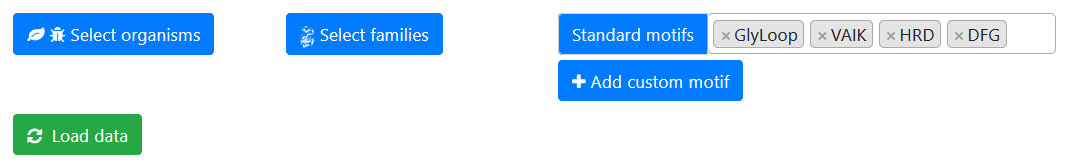
Database searching is dynamic and can be limited by organism and kinase families before the
data are loaded, and refined at any time afterwards. Select motifs of interest
from the dropdown list and click the green "Load table" button to initiate the search and load the results table.
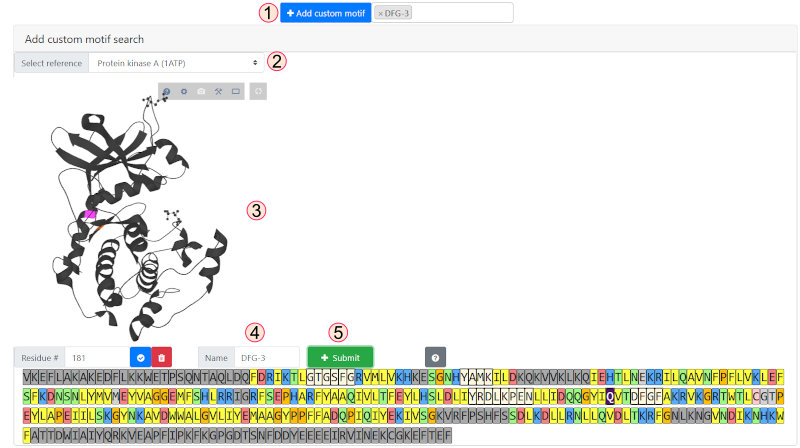
Custom positions can be added to the filter by clicking on the custom motif button and selecting a reference
kinase from the dropdown. The residue position
can be selected by residue or by clicking on the structure or on the kinase sequence. Dark gray sequence regions are outside of the core kinase domain and unavailable
for selection. Once a position is selected, give your motif a name (here we have selected the DFG-3 position, which is now highlighted dark purple in the sequence)
and click "Submit."
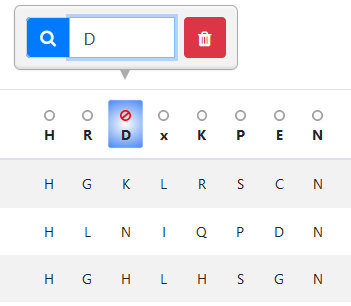
To activate a filter, click on the position of interest in the table header. Enter the residue(s) you wish to
add to the filter using single-letter amino
acid code. You can clear a filter by clicking the red trash icon. To invert the selection, click the circle above the position of interest, which will become
a "not" sign as in the image. All filters can be reset and the data reloaded by clicking the "Reset table" button. You can also add a text search using the
"Search" input in the table toolbar. Table results (those currently loaded on the page) can be downloaded using the gray button at the table's top right.
Viewing kinase alignments
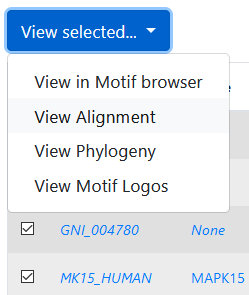
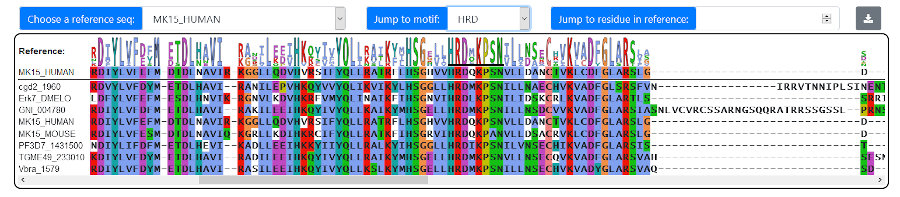
Alignments can be visualized in a custom in-browser viewer. To do so, first select kinases either from the Family
or Motif browsers by clicking the checkboxes, or from the
phylogeny viewer (by clicking the kinase names), then select
"View alignment" from the "View selected…" drop-down menu. The alignment may take a moment to render; please note that it is not recommended that
you attempt to view >100-150 sequences in the in-browser viewer, as your browser may face performance issues. Once the alignment is loaded, you will have the
option to select a reference sequence. The consensus sequence logo is rendered above each position in the alignment. You can scan through the alignment
by using the scrollbar, by entering a residue number (of the reference sequence), or by using the "Jump to motif" selection, which will also underline
the respective motif in the consensus region.
Note that the sub-alignments can be downloaded by clicking the gray download button in the toolbar.
Using the kinase phylogenetic tree viewer
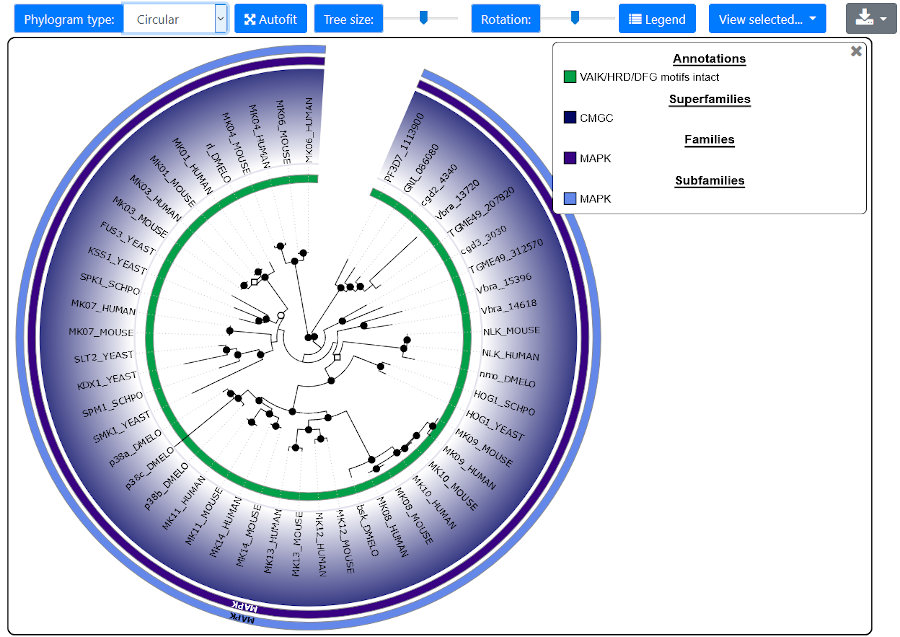
Two styles of phylogenetic trees are available for render: rectangular and circular. Note that trees
containing hundreds of elements may be
time-consuming to render, especially for circular trees. The tree size relative to the annotation may be adjusted with the corresponding slider.
Tree height (for rectangular) or rotation (for circular) are also adjustable using sliders. Colors can be customized by clicking on the
corresponding swatch in the legend. The raw trees (in phyloXML format) or the graphics (in SVG format) are downloadable by clicking on the
Download button.
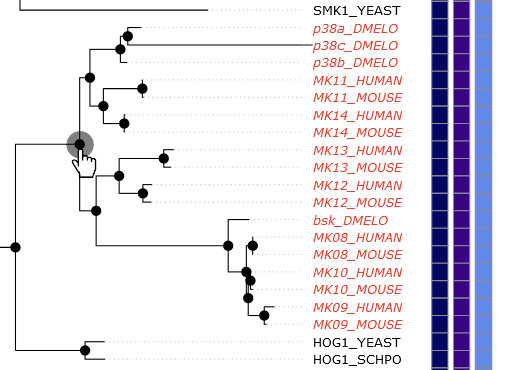
To select kinases in the tree, you can either click on individual gene names or click on the branch
points in the tree. When you hover over a name, a red circle will appear over the end of the corresponding branch (its leaf). When you hover
your mouse over a branch point, all kinases that are child leaves of that point will turn red (see image). Once selected, the kinase names
will be bold. Hovering over kinase names will pop-up information about the kinase, including links to its kinase page and UniProt entry.
Viewing kinase details and accessing available structures

Each kinase in the database has a page giving details on its motifs and known functional properties.
For model organisms, a precis of the literature has been downloaded from UniProt (under CC-BY), along with links to the first 10 references. For
non-model organisms, we have attempted to concisely review available literature on a given kinase in a few sentences, focusing on functional
information rather than inhibitors that may have been tested or structures that have been solved without additional biological data. Note that
for proteins with multiple kinase domains, both domains are detailed on a single page. An overview of the domain architecture (collected from the
SMART database) is presented in a cartoon form. Hovering over domains gives details.
Links to structural information are present, when available – this is defined as deposited structures in which one chain has >95% identity
with the kinase domain in question. Currently, this list is limited to the first 10 structures identified. In the future we hope to make homology
models available for all kinases in the database for which high quality models can be generated.
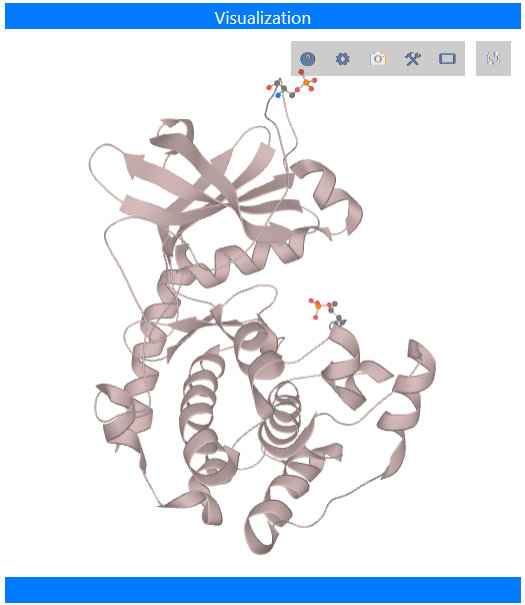
Two structural views are available: the kinase chain only, or the entire biological assembly, as defined
in the PDB. This may include additional proteins, ions, small molecules (inhibitors or crystallographic reagents), and solvent. Drag with the
left mouse button to rotate the model, or with the right button to zoom in or out. Dragging with the middle mouse button moves the model. See the
LiteMol documentation for more information. The goal of LiteMol is simple viewing of structures
within a web browser; for more complex manipulations, we recommend downloading the file from the Protein Data Bank
and viewing in your favorite program, perhaps PyMOL or
UCSF Chimera.